den 16 januari 2019
EU-kommissionen godkänner Symkevi
Den Europeiska kommissionen har godkänt Symkevi (tezakaftor/ivakaftor) i kombination med ivakaftor för patienter med cystisk fibros äldre än 12 år med vissa specifika mutationer i CFTR-genen

Lena Hjelte
Professor Stockholm CF center.
Vertex Pharmaceuticals meddelar att den Europeiska kommissionen har gett ett marknadsföringsgodkännande för tezakaftor/ivakaftor, att användas i kombination med ivakaftor för behandling av cystisk fibros (CF) hos personer äldre än 12 år, som antingen har två kopior av F508del-mutationen i CFTR-genen, eller en kopia av F508del-mutationen och en kopia av en av följande 14 mutationer där CFTR-proteinet visar kvarvarande aktivitet: P67L, R117C, L206W, R352Q, A455E, D579G, 711+3A→G, S945L, S977F, R1070W, D1152H, 2789+5G→A, 3272-26A→G, and 3849+10kbC→T.
Godkännandet av Symkevi i kombination med ivacaftor är positiva nyheter för patienter i Sverige med cystisk fibros. Genom godkännandet har vi nu ytterligare ett behandlingsalternativ tillgängligt för patienter med den vanligaste mutationen F508del i dubbel upplaga utöver Orkambi. Med Symkevi godkänt får vi även möjligheten att behandla ytterligare en patientgrupp, det vill säga patienter med viss kvarstående funktion i CFTR proteinet i kombination med mutationen F508del. Dessa nya läkemedel är mutationsspecifika och behandlar den underliggande proteindefekten, säger professor Lena Hjelte Stockholm CF center vid Karolinska Universitetssjukhuset i Huddinge. Den Europeiska läkemedelsmyndighetens vetenskapliga kommitté för särläkemedel (Commitee for Orphan Medicinal Products) har dessutom nyligen rekommenderat bibehållen särläkemedelsklassificering för tezakaftor/ivakaftor i kombination med ivakaftor (1).
Den Europeiska kommissionens godkännande baserade sig på resultaten från två pivotala Fas III-studier, EVOLVE och EXPAND, vilka publicerats i den medicinska tidskriften the New England Journal of Medicine, i november 2017 (2, 3). Resultaten visade att behandling med tezakaftor/ivakaftor i kombination med ivakaftor gav fördelar hos olika patientgrupper med CF, inklusive statistiskt signifikant förbättring i lungfunktionen mätt som den absoluta förändringen i ppFEV1 från ursprungsvärdet, en generellt väl tolererad säkerhetsprofil samt ingen ökning av andningsrelaterade biverkningar jämfört med placebo.
Förbättringarna i lungfunktionen visade en medelförändring av den absoluta förändringen av ppFEV1 jämfört med placebo på 4,0 procentenheter (p<0,0001) och 6,8 procentenheter (p<0,0001) i EVOLVE- respektive EXPAND-studien. De vanligaste biverkningarna hos patienter som fått behandling med tezakaftor/ivakaftor i kombination med ivakaftor var huvudvärk och nasofaryngit.
Godkännandet är en viktig milstolpe för patienter med cystisk fibros i Sverige. Framförallt för den grupp som hittills inte haft något tillgängligt behandlingsalternativ för den CFTR-protein defekt som är orsaken till deras sjukdom. Med dagens EU-godkännande rör vi oss snabbt mot att kunna behandla 90 procent av alla patienter med cystisk fibros. Vi ser fram emot att avsluta trepartsförhandlingarna med TLV och landstingen rörande tezakaftor/ivakaftor inom en snar framtid, för att säkerställa att patienter i Sverige kan få tillgång till precisionsmedicin mot cystisk fibros så snart som möjligt, säger Morgan Savage, Nordisk regionchef på läkemedelsföretaget Vertex. Tezakaftor/ivakaftor i kombination med ivakaftor godkändes av FDA (U.S. Food and Drug Administration) i februari 2018 och av Health Canada i juni 2018 (4, 5).
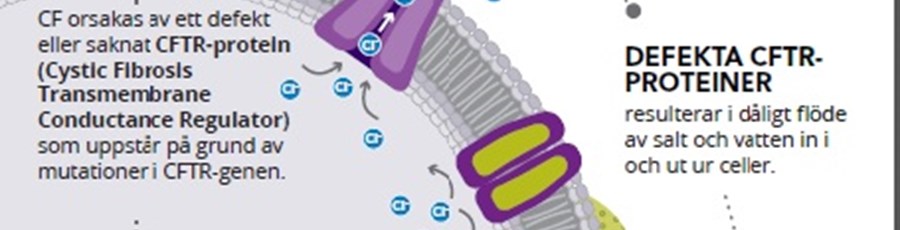
Cystisk fibros (CF) är en sällsynt genetisk sjukdom som förkortar livet och drabbar cirka 75 000 personer i Nordamerika, Europa och Australien (6). I Sverige föds cirka 20 barn med CF årligen och cirka 670 svenskar är drabbade av sjukdomen (12). CF orsakas av brist på eller defekt CFTR-protein, till följd av mutationer i CFTR-genen (7). Barn måste ärva två defekta CFTR-gener, en från varje förälder, för att få CF (8). Det finns cirka 2 000 kända mutationer i CFTR-genen (9). Vissa av dessa mutationer, vilka kan bestämmas med hjälp av ett genetiskt eller genotyp-test, leder till CF genom att de orsakar för få eller icke fungerande CFTR-proteiner vid cellytan (7, 8). Den defekta funktionen eller avsaknaden av CFTR-protein leder till ett sämre flöde av salt och vatten in och ut ur cellerna hos ett antal organ i kroppen. I lungorna leder det till en ansamling av onormalt tjockt och segt slem som kan orsaka kroniska lunginfektioner och fortskridande lungskador hos många patienter samt leda till en för tidig död. För personer med CF är medianåldern för dödsfall mitten till slutet av tjugoårsåldern (10).
Om Symkevi (tezakaftor/ivakaftor) och ivakaftor Vissa mutationer leder till ett CFTR-protein som inte har processats i cellen på ett normalt sätt och därmed inte når cellens yta. Tezacaftor är ett läkemedel som riktar in sig på att korrigera denna processdefekt i CFTR-proteinet för att möjliggöra att det når cellytan, där sedan Ivacaftor fungerar genom att öppna befintliga jonkanaler och på så sätt förbättra funktionen av proteinet.
För fullständig produktinformation, se www.ema.europa.eu
Om EVOLVE- och EXPAND-studierna Resultaten från de två fas III-studierna EVOLVE och EXPAND publicerades i den medicinska tidskriften New England Journal of Medicine i november 2017 (2, 3) och omfattade cirka 750 personer med CF äldre än 12 år med två kopior av F508del-mutationen eller en kopia av F508del-mutationen och en andra mutation som är kopplad till kvarvarande aktivitet hos CFTR-proteinet.
Båda studierna visade att patienter behandlade med tezakaftor/ivakaftor i kombination med ivakaftor fick en statistiskt signifikant förbättring i lungfunktionen, mätt som den absoluta förändringen i ppFEV1 från ursprungsvärdet.
Behandlingen var generellt väl tolererad och de vanligaste biverkningarna (≥10%) hos de patienter som fått tezackaftor/ivakaftor i kombination med ivakaftor var huvudvärk (14% jämfört med 12% i placebo-gruppen) samt nasofaryngit (12% jämfört med 10% i placebo-gruppen) (1).
Om särläkemedel
Europeiska läkemedelsmyndighetens vetenskapliga kommitté för särläkemedel (Commitee for Orphan Medicinal Products) kan bevilja särläkemedelsstatus för läkemedel som uppfyller ett otillfredställt medicinskt behov eller innebär en betydande fördel för patienter med en livshotande eller kroniskt funktionsnedsättande sjukdom, som drabbar ett litet antal patienter (11).
Referenser
1. EMA, COMP meeting report on the review of applications for orphan designation: September 2018. Available at : https://www.ema.europa.eu/documents/committee-report/comp-meeting-report-review-applications-orphan-designation-september-2018_en.pdf. Last accessed: October 2018
2. Taylor-Cousar, J.L., Munck, A., McKone E.F. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. New England Journal of Medicine. 2017. Vol. 377:2013-2023
3. Rowe, S.M, Daines, C, Ringshausen, F.C. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. New England Journal of Medicine. 2017. Vol.377:2024-2035
4. FDA. Approved Drug Letter. SYMDEKO (tezacaftor/ivacaftor) Tablets 100/150 mg. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210491Orig1s000Approv.pdf. Last accessed:
October 2018
5. Health Canada. Drug product Database. SYMDEKO. Available at: https://health-products.canada.ca/dpd-bdpp/dispatch-repartition.do. Last accessed: October 2018
6. Jabar.A. New and Evolving Therapies for Cystic Fibrosis Patients. Pediatric allergy, Immunology, and pulmonology. Vol 27, No. 2 7. Cystic Fibrosis Foundation. Basics of The CFTR Protein. Available at: https://www.cff.org/Research/Research-Into-the-Disease/Basics-of-the-CFTR-Protein/. Last accessed: October 2018
8. NHS choices. Cystic Fibrosis. Available at: https://www.nhs.uk/conditions/cystic-fibrosis/Last accessed: October 2018
9. Quon, B.S, Rowe, M.R. New and Emerging Targeted Therapies for Cystic Fibrosis. The British Medical Journal. 2016. 352: i859
10. European Cystic Fibrosis Society Patient Registry Annual Data Report 2015. Available at: https://www.ecfs.eu/news/ecfs-patient-registry-2015-annual-report. Last accessed: October 2018 11. EMA. Committee for Orphan Medicinal Products (COMP). Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/general/general_content_000263.jsp&mid=WC0b01ac0580028e30. Last accessed: October 2018
12. Socialstyrelsen, Ovanliga diagnoser, Cystisk fibros, 2016-06-21 https://www.socialstyrelsen.se/ovanligadiagnoser/cystiskfibros#anchor_2




Annonser
.gif")


")

