den 26 februari 2025
Ny metod söker igenom 10 sextiljoner läkemedelsmolekyler
En färsk studie visar att datoralgoritmer kan användas för att hitta potenta molekyler som skulle kunna utvecklas till läkemedel mot inflammation. I artikeln presenterar forskarna också hur samma strategi kan användas för att söka igenom 10 sextiljoner alternativ för att hitta den bästa läkemedelskandidaten.

Jens Carlsson
Professor. Foto: Mikael Wallerstedt.
En av de största utmaningarna inom läkemedelsutveckling är att hitta rätt kandidat bland ett enormt antal potentiella molekyler. En ny studie, som publicerats i Nature Communications, visar att det går att hitta läkemedelsmolekyler genom att modellera dem med hjälp av datoralgoritmer.
− Vi utnyttjar datormodellerna för att söka igenom databaser med flera miljarder molekyler. Metoden kommer att kunna snabba upp den kostnadskrävande läkemedelsutvecklingsprocessen, säger Jens Carlsson, en av författarna till studierna.
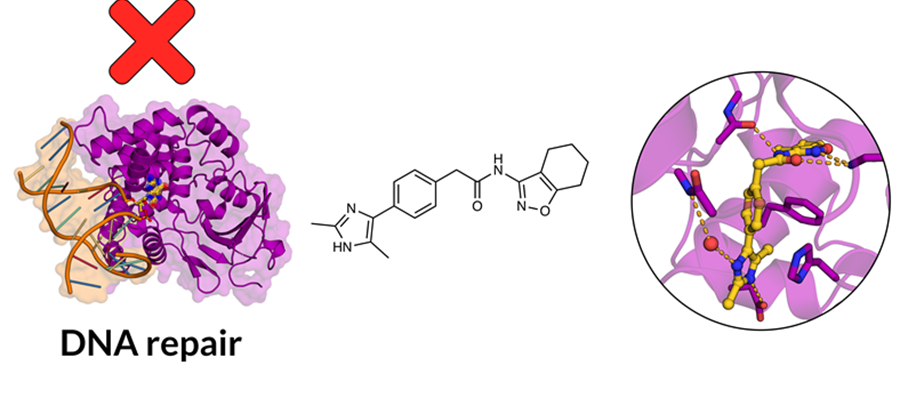
Läkemedelspotential för inflammation
I den aktuella studien, som är ett samarbete med Karolinska institutet och Stockholms universitet, arbetar man med enzymet OG1, ett protein som reparerar skador på cellernas DNA. Forskarna var intresserade av att hitta en molekyl som kunde binda till detta målprotein och på så sätt påverka enzymaktiviteten. Med hjälp av modeller av proteindesignade fler än hundra olika molekyler som sedan tillverkas. När molekylerna testades mot enzymet kunde forskarna se att de hämmade dess aktivitet och hade antiinflammatorisk effekt.
− Det är fantastiskt att vi idag kan designa molekyler och visa att de faktiskt fungerar exakt som vi hoppar. Samma strategi kommer att fungera för många andra proteiner och sjukdomar, säger Jens Carlsson.
Börjar med enkla molekyler
Tidigare har läkemedelsutveckling fokus på att screena stora kemiska bibliotek som innehåller läkemedelsliknande molekyler. Denna metod är dock mycket kostnadskrävande och misslyckas ofta att hitta bra startpunkter för läkemedelsutveckling. I den aktuella studien har forskarna i stället använt sig av så kallad fragmentbaserad läkemedelsdesign. Metoden innebär att man först identifierar en mycket liten molekyl som kan binda till målproteinet. När en sådan molekyl har hittats, så kan forskarna stegvis vidareutveckla den för att skapa ett läkemedel.
Det är som att lägga ett pussel. Vi startar med en pusselbit och bygger sedan upp en successivt läkemedelsmolekyl genom att addera nya bitar. Till slut har vi en läkemedelsmolekyl som passar perfekt i målproteinet, säger Jens Carlsson.
I studien har undersökningarna använt sig ett företag som tillverkar molekyler på beställning. De har sedan skapat ett datorprogram som kan söka igenom alla miljarder molekyler som idag finns att köpa. Med hjälp av superdatorer kan utforska vilka molekyler som kan binda till målproteinets yta.
− Vi letade först bland miljarderna, där kunde vi snabbt få molekylerna tillverkade och testade. Och det gick bra, vi hittade molekyler som fungerade. Mot slutet av studien började vi tänka - hur långt kan vi gå, om vi inte har kravet att vi måste kunna köpa dem?
Doktoranden Andreas Luttens skrev då ett nytt datorprogram som kunde generera alla möjliga molekyler. Antalet potentiella alternativ landade då på 10 sextiljoner (10 22 eller en etta följt av 22 nollor). Forskarna kunde sedan visa att samma metod som de används för att söka bland molekylerna som finns att köpa, kommer också att fungera för att söka bland de 10 sextiljonerna.
− Med vår strategi kan vi söka igenom 10 sextiljoner läkemedelsmolekyler på ett effektivt sätt. Vi kommer inom en snar framtid att kunna testa alla möjliga läkemedelsmolekyler i våra datormodeller, ett genombrott som har stor potential.
Ställer nya krav på läkemedelsskemiterna
Även om det nu finns beräkningskraft att utforska ett enormt antal molekyler i jakt på nya läkemedel är det inte alltid säkert att de nya substanserna faktiskt går att tillverka.
− Framtiden kommer att kräva utveckling av nya tillverkningsmetoder för att lyckas med molekylerna som datorberäkningarna så snabbt kan designa, säger Jens Carlsson.
Källa: Uppsala universitet.




Annonser
.gif")


")

